![]()
Skill:
• Analysis of cladograms to deduce evolutionary relationships
Constructed cladograms all typically share certain key features:
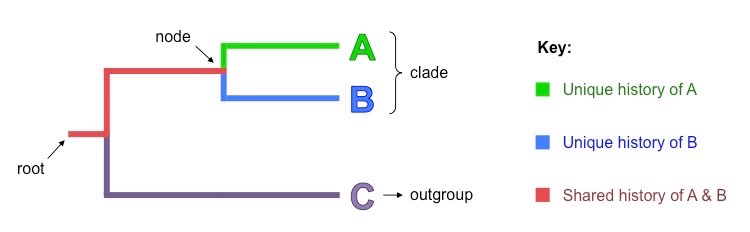
- Root – The initial ancestor common to all organisms within the cladogram (incoming line shows it originates from a larger clade)
- Nodes – Each node corresponds to a hypothetical common ancestor that speciated to give rise to two (or more) daughter taxa
- Outgroup – The most distantly related species in the cladogram which functions as a point of comparison and reference group
- Clades – A common ancestor and all of its descendants (i.e. a node and all of its connected branches)
Key Features of a Cladogram
Constructing Cladograms
Cladograms can be constructed based on either a comparison of morphological (structural) features or molecular evidence
- Historically, structural features were used to construct cladograms, but molecular evidence is now more commonly used
1. Using Structural Evidence
Step 1: Organise selected organisms according to defined characteristics
- Use characteristics that are developmentally fixed (i.e. innate) and not influenced by environmental pressures
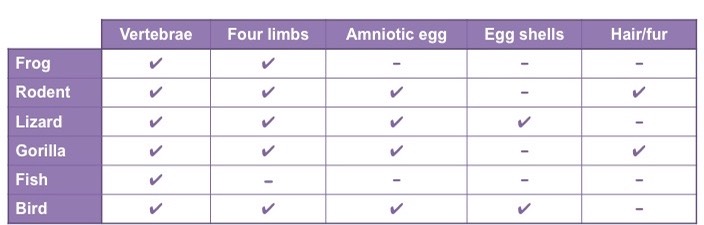
Step 2: Sequentially order organisms according to shared characteristics to construct a cladogram
- Grouping of organisms may be facilitated by constructing a Venn diagram prior to developing a cladogram
- Each characteristic will be represented by a node, with more common characteristics representing earlier nodes
- The species with the least number of characteristics in common will represent the outgroup (establishes baseline properties)

2. Using Molecular Evidence
Step 1: Select a gene or protein common to a range of selected organisms
- Examples of molecules which are ubiquitously found in many animals include haemoglobin and cytochrome c
Step 2: Copy the molecular sequence (DNA or amino acid) for each of the selected organisms
- Use online databases such as Genbank or Ensembl to identify relevant DNA or amino acid sequences
- Sequences can be collated in a Word document and then saved as a document in plain text format (.txt)
- Before each sequence, designate a species name preceded by a forward arrow (e.g. '>Human’ or ‘>Chimpanzee’)
Step 3: Run a multiple alignment to compare molecular sequences (DNA or amino acid)
- Multiple alignment software compares DNA or protein sequences for similarities and differences
- Closely related species are expected to have a higher degree of similarity in their molecular sequence
- Clustal Omega is a free online tool that will align multiple DNA or amino acid sequences for comparison
Step 4: Generate a phylogeny tree (cladogram) from multiple alignment data
- Clustal Omega can generate branched phylograms after a sequence alignment is completed (select ‘Phylogenetic Tree’)
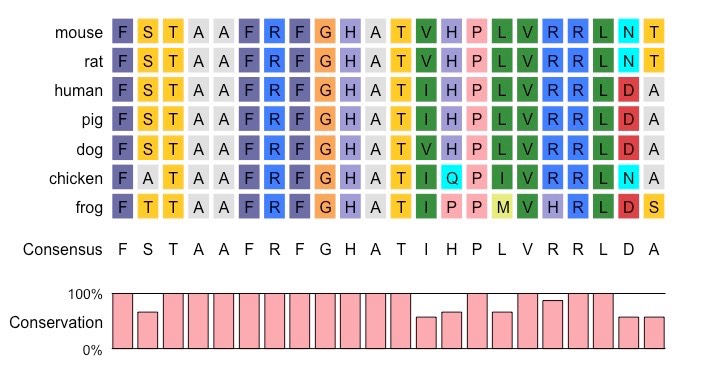
- Below is a plain text file that can be uploaded to compare amino acid sequences from different species:
- HBA – Haemoglobin alpha chain (amino acid sequence) from various species
Multiple Alignment of a Protein Sequence from Various Species