
![]()
Application:
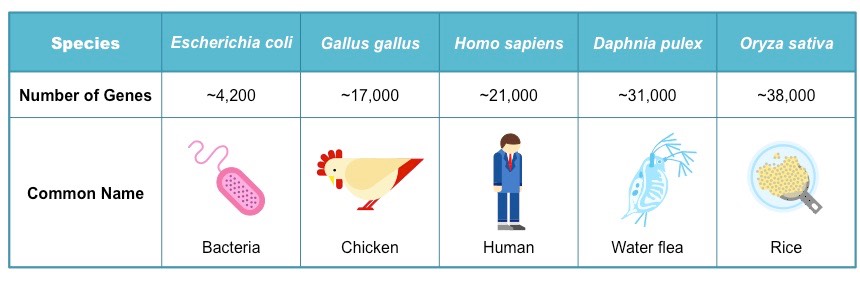
• Comparison of the number of genes in humans with other species
The number of genes present in an organism will differ between species and is not a valid indicator of biological complexity
The number of genes in a genome is usually predicted by identifying sequences common to genes
- These identifying regions may include expressed sequence tags (ESTs) or sequences that are homologous to known genes
- The presence of pseudogenes and transposons make accurate counts of unique gene numbers very difficult
As scientists may use different approaches to predicting gene numbers, final estimations can vary significantly
- For instance, the number of genes in rice (Oryza sativa) is estimated as being between 32,000 – 50,000
- The number of genes in humans is estimated as being between 19,000 – 25,000
Gene Comparisons Between Different Species
![]()
Skill:
• Use of a database to determine differences in the base sequence of a gene in two species
Gene sequences from different species can be identified and then compared using two online resources:
- GenBank – a genetic database that serves as an annotated collection of DNA sequences
- Clustal Omega – an alignment program that compares multiple sequences of DNA


Identifying Gene Sequences
GenBank can be used to identify the DNA sequence for a gene in a number of different species
To identify a specific gene sequence:
- Change the search parameter from nucleotide to gene and type in the name of the gene of interest
- Choose the species of interest and click on the link (under ‘Name / Gene ID’)
- Scroll to the ‘Genomic regions, transcripts and products’ section and click on the ‘FASTA’ link
Below are examples of different genes that may be searched for:
- HBB – Haemoglobin beta gene
- COX1 – Cytochrome oxidase 1 gene
- F8 – Coagulation factor VIII gene
- IGF1R – Insulin growth factor 1 receptor gene
Comparing Gene Sequences
Clustal Omega aligns multiple gene sequences to allow for the determination of differences in the base sequence
To construct a multiple alignment:
- Change the input sequence type to DNA and paste the relevant FASTA sequences into the provided space
- Before each sequence designate a species name preceded by a forward arrow (e.g. '>Human’ or ‘>Chimpanzee’)
- Alternatively, sequences can be saved as a document in plain text format (.txt) and then uploaded
- When all sequences have been included, click ‘Submit’ (under step 3)
Clustal Omega possesses several useful features that are absent in alternative tools like BLASTN:
- Multiple (more than two) sequences can be compared at once
- Sequence consensus is colour coded in a Jalview applet found under ‘Result Summary’ (requires Java)
- Branched phylograms can be generated to show evolutionary relationships (under ‘Phylogenetic Tree’)
Below is a plain text file that can be uploaded to compare gene sequence fragments from different species:
- CHRNE – Cholinergic receptor epsilon (nicotinic) for various species
Sequence Alignment of DNA from Two Species
